Monday, March 19. 2012
Mess With MIT's Enormous Homemade 1970s Synthesizer
Via The Atlantic
-----
Joe Paradiso, an associate professor at MIT's Media Lab and director of the Responsive Environments group, started building this analog music synthesizer in 1973. Now, it streams music live from the MIT Museum, and users can manipulate it remotely via the web. With ten people at a time fiddling with the machine's knobs and switches (literally -- the web interface controls the synth's motorized parts), the music often sounds a little like a chorus of baby UFOs getting rowdy. The chaos of collective control is pure fun -- dial down the tempo to turn the bleeps and echoes to syrup, only to see someone dial it way up to frenetic levels again. Someone hits the "slapback drum machine" button just as you hit "growl + gliding & rumbling," and texture builds on texture. Visit http://synth.media.mit.edu/patchwerk/ and enter a username to get in line to participate.
This video was produced by Lucy Lindsey and Melanie Gonick at MIT. Lindsey describes the decades-long project in an article for the MIT New Office:
It was a time, [Paradiso] says, when information and parts for do-it-yourself projects were scarce, and digital synthesizer production was on the rise. But, he decided to tackle the project — without any formal training ... Paradiso gathered information from manufacturers’ data sheets and hobbyist magazines he found in public libraries. He taught himself basic electronics, scrounged for parts from surplus stores and spent a decade and a half building modules and hacking consumer keyboards to create the synth, which he completed in the 1980s.
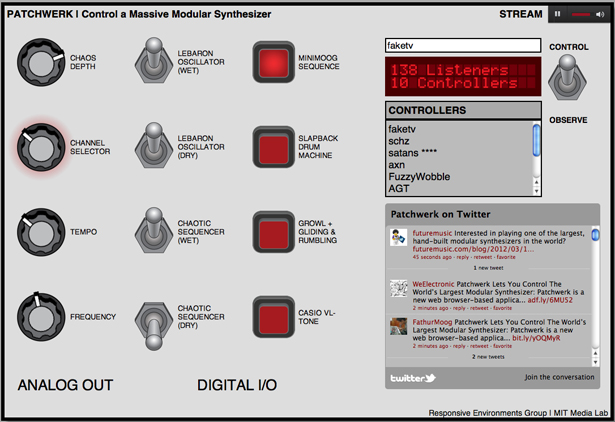
The Patchwerk interface
For more information about the Paradiso Synthesizer, visit http://web.media.mit.edu/~joep/synth.html.
Friday, February 24. 2012
USB stick can sequence DNA in seconds
Via NewScientist
-----
</i>")
(Image: Oxford Nanopore Technologies)
It may look like an ordinary USB memory stick, but a little gadget that can sequence DNA while plugged into your laptop could have far-reaching effects on medicine and genetic research.
The UK firm Oxford Nanopore built the device, called MinION, and claims it can sequence simple genomes – like those of some viruses and bacteria – in a matter of seconds. More complex genomes would take longer, but MinION could also be useful for obtaining quick results in sequencing DNA from cells in a biopsy to look for cancer, for example, or to determine the genetic identity of bone fragments at an archaeological dig.
The company demonstrated today at the Advances in Genome Biology and Technology (AGBT) conference in Marco Island, Florida, that MinION has sequenced a simple virus called Phi X, which contains 5000 genetic base pairs.
Proof of principle
This is merely a proof of principle – "Phi X was the first DNA genome to be sequenced ever," says Nick Loman, a bioinformatician at the Pallen research group at the University of Birmingham, UK, and author of the blog Pathogens: Genes and Genomes. But it shows for the first time that this technology works, he says. "If you can sequence this genome you should be able to sequence larger genomes."
Oxford Nanopore is also building a larger device, GridION, for lab use. Both GridION and MinION operate using the same technology: DNA is added to a solution containing enzymes that bind to the end of each strand. When a current is applied across the solution these enzymes and DNA are drawn to hundreds of wells in a membrane at the bottom of the solution, each just 10 micrometres in diameter.
Within each well is a modified version of the protein alpha hemolysin (AHL), which has a hollow tube just 10 nanometres wide at its core. As the DNA is drawn to the pore the enzyme attaches itself to the AHL and begins to unzip the DNA, threading one strand of the double helix through the pore. The unique electrical characteristics of each base disrupt the current flowing through each pore, enough to determine which of the four bases is passing through it. Each disruption is read by the device, like a tickertape reader.
Long strands, and simple
This approach has two key advantages over other sequencing techniques: first, the DNA does not need to be amplified - a time-consuming process that replicates the DNA in a sample to make it abundant enough to make a reliable measurement.
Second, the devices can sequence DNA strands as long as 10,000 bases continuously, whereas most other techniques require the DNA to be sheared into smaller fragments of at most a few hundred bases. This means that once they have been read they have to be painstakingly reassembled by software like pieces of a jigsaw. "We just read the entire thing in one go," as with Phi X, says Clive Brown, Oxford Nanopore's chief technology officer.
But Oxford Nanopore will face stiff competition. Jonathan Rothberg, a scientist and entrepreneur who founded rival firm 454 Life Sciences, also announced at the AGBT conference that his start-up company, Ion Torrent, will be launching a desktop sequencing machine. Dubbed the Ion Proton, it identifies bases by using transistors to detect hydrogen ions as they are given off during the polymerisation of DNA.
This device will be capable of sequencing a human genome in 2 hours for around $1000, Rothberg claims. Nanopores are an "elegant" technology, he says, but Ion Torrent already has a foot in the door. "As we saw last summer with the E. coli outbreak in Germany, people are already now using it," he says.
Pocketful of DNA
By contrast, the MinION would take about 6 hours to complete a human genome, Brown claims, though the company plans to market the device for use in shorter sequencing tasks like identifying pathogens, or screening for genetic mutations that can increase risk of certain diseases. Each unit is expected to cost $900 when it goes on sale later this year.
"The biggest strength of nanopore sequencing is that it generates very long reads, which has been a limitation for most other technologies," says Loman. If the costs, quality, ease of use and throughput can be brought in line with other instruments, it will be a "killer technology" for sequencing, he says.
As for clinical applications, David Rasko at the Institute for Genome Sciences at the University of Maryland in Baltimore, says the MinION could have huge benefits. "It may have serious implications for public health and it could really change the way we do medicine," he says. "You can see every physician walking around the hospital with a pocketful of these things." And it will likely increase the number of scientists generating sequencing data by making the technology cheaper and more accessible, he says.
Thursday, February 23. 2012
Alan Turing's reading list (with readable links)
MOTHER artificial intelligence forces nerds to do the chores… or else
Via Slash Gear
-----
If you’ve ever been inside a dormitory full of computer science undergraduates, you know what horrors come of young men free of responsibility. To help combat the lack of homemaking skills in nerds everywhere, a group of them banded together to create MOTHER, a combination of home automation, basic artificial intelligence and gentle nagging designed to keep a domicile running at peak efficiency. And also possibly kill an entire crew of space truckers if they should come in contact with a xenomorphic alien – but that code module hasn’t been installed yet.

The project comes from the LVL1 Hackerspace, a group of like-minded
programmers and engineers. The aim is to create an AI suited for a home
environment that detect issues and gets its users (i.e. the people living in
the home) to fix it. Through an array of digital sensors, MOTHER knows
when the trash needs to be taken out, when the door is left unlocked, et
cetera. If something isn’t done soon enough, she it can even
disable the Internet connection for individual computers. MOTHER can
notify users of tasks that need to be completed through a standard
computer, phones or email, or stock ticker-like displays. In addition,
MOTHER can use video and audio tools to recognize individual users,
adjust the lighting, video or audio to their tastes, and generally keep
users informed and creeped out at the same time.
MOTHER’s abilities are technically limitless – since it’s all based on open source software, those with the skill, inclination and hardware components can add functions at any time. Some of the more humorous additions already in the project include an instant dubstep command. You can build your own MOTHER (boy, there’s a sentence I never thought I’d be writing) by reading through the official Wiki and assembling the right software, sensors, servers and the like. Or you could just invite your mom over and take your lumps. Your choice.
Wednesday, February 22. 2012
A Wirelessly Controlled Pharmacy Dispenses Drugs From Within Your Abdomen
Via PopSci
-----

In the future, implantable computerized dispensaries will replace trips to the pharmacy or doctor’s office, automatically leaching drugs into the blood from medical devices embedded in our bodies. These small wireless chips promise to reduce pain and inconvenience, and they’ll ensure that patients get exactly the amount of drugs they need, all at the push of a button.
In a new study involving women with osteoporosis, a wirelessly controlled implantable microchip successfully delivered a daily drug regimen, working just as well, if not better, than a daily injection. It could be an elegant solution for countless people on long-term prescription medicines, researchers say. Patients won’t have to remember to take their medicine, and doctors will be able to adjust doses with a simple phone call or computer command.
Pharmacies-on-a-chip could someday dispense a whole suite of drugs, at pre-programmed doses and at specific times, said Robert Langer, the Institute Professor at the David H. Koch Institute for Integrative Cancer Research at MIT, who is a co-author on the study.
“It really depends on how potent the drugs are,” he said. “There are a number of drugs for things like multiple sclerosis, cancer, and some vaccines that would be potent enough.”
Langer and fellow MIT professor Michael Cima developed an early version of an implantable drug-delivery chip in the late 1990s. They co-founded a company called MicroCHIPS Inc., which administered the study being published today in Science Translational Medicine. The team decided to work with osteoporosis patients because the disease, and the drug used to treat it, presented a series of special opportunities, Langer said. A widely used drug called teriparatide can reverse bone loss in people with severe osteoporosis, but it requires a daily injection to work properly. This means up to 75 percent of patients give up on the therapy, Langer said. It’s also a very potent drug that requires microgram doses, making it an ideal candidate for a long-term dispensary implant.
Getting the chips to work well required some tinkering on the part of the company, including the addition of a hermetic seal and drug-release system that can work in living tissue. The chip contains a cluster of tiny wells, about the size of a pinprick, which store the drug. Each well is sealed with an ultrathin layer of platinum and titanium, Langer said. At programmed times or at the patient’s command, an external radio-frequency device sends a signal to the chip, which applies a voltage to the metal film, melting it and releasing the drug. The wells melt one at a time.
“It’s like blowing a fuse, the way we’ve got it set,” Langer said. He said the amount of metal is near nanoscale levels and is not toxic.
The team also had to ensure the chips were secure and could not be hacked. The chips communicate via a special frequency called the Medical Implant Communications Service band, approved by both the FCC and the FDA. A bidirectional communications link between the chip and a receiver enables the upload of implant status information, including confirmation of dose delivery and battery life. A patient or doctor would then enter a special code to administer or change the dose, Langer said.
The research team recruited seven women in Denmark who had severe osteoporosis and surgically implanted the chips into their abdomens in January 2011. The chips stored 20 doses of the drug. The patients had the implants for a year, and they proved extremely popular, Langer said. “They didn’t think about the fact that they had it, since they didn’t have to have injections,” he said.
Ultimately, the device delivered dosages comparable to daily injections, and there were no negative side effects. There was no skipping the shot if a patient didn’t feel like visiting the doctor — complying with a prescription is of key importance, said Cima, the David H. Koch Professor of Engineering at MIT. “This avoids the compliance issue completely, and points to a future where you have fully automated drug regimens,” he said.
The study points out one interesting phenomenon that will inform future research and development on these types of implants. When you implant a device into a person’s body, the body forms a fibrous, collagen-based membrane that surrounds the foreign device. This can affect how well drugs can move from the device and into the body, which in turn affects dosage requirements and pharmaceutical potency. One of the aims of this study was to examine the effects of that collagen membrane, and the researchers found it did not have any deleterious effects on the drug.
Now that these chips have been proven to work, Langer and the others want to test them with other drugs and for longer dosage periods, he said. Because the well caps melt one at a time, the chips could be used to deliver different types of drugs, even those that would normally interact with each other if taken in shot or pill form, he said. The team wants to build a version with 365 doses to see how well it works.
It could even be used as a long-term sensing device, he said, an interesting possibility of its own. Medical sensing implants can degrade once they’re in the body, so implants that could check for things like blood sugar or cancer antibodies can lose their effectiveness. But a chip with multiple sensors can work a lot longer — once a sensor is befouled, simply melt another well and expose a fresh one, Langer said.
The ultimate goal is to create a chip that could combine sensing and drug delivery — an implantable diagnostic machine that can deliver its own therapy.
“Someday it would be great to combine everything, but that will obviously take longer,” Langer said.
Monday, February 20. 2012
Envelope for an artificial cell
Via PhysOrg
-----

Neal Davaraj watches as undergraduate student Weilong Li works on a next
step in their quest to create an entirely artificial cell.
Chemists have taken an important step in making artificial life forms from scratch. Using a novel chemical reaction, they have created self-assembling cell membranes, the structural envelopes that contain and support the reactions required for life.
Neal Devaraj, assistant professor of chemistry at the University of California, San Diego, and Itay Budin, a graduate student at Harvard University, report their success in the Journal of the American Chemical Society.
“One of our long term, very ambitious goals is to try to make an artificial cell, a synthetic living unit from the bottom up – to make a living organism from non-living molecules that have never been through or touched a living organism,” Devaraj said. “Presumably this occurred at some point in the past. Otherwise life wouldn’t exist.”
By assembling an essential component of earthly life with no biological precursors, they hope to illuminate life’s origins.
“We don’t understand this really fundamental step in our existence, which is how non-living matter went to living matter,” Devaraj said. “So this is a really ripe area to try to understand what knowledge we lack about how that transition might have occurred. That could teach us a lot – even the basic chemical, biological principles that are necessary for life.”
Molecules that make up cell membranes have heads that mix easily with water and tails that repel it. In water, they form a double layer with heads out and tails in, a barrier that sequesters the contents of the cell.
Devaraj and Budin created similar molecules with a novel reaction that joins two chains of lipids. Nature uses complex enzymes that are themselves embedded in membranes to accomplish this, making it hard to understand how the very first membranes came to be.
“In our system, we use a sort of primitive catalyst, a very simple metal ion,” Devaraj said. “The reaction itself is completely artificial. There’s no biological equivalent of this chemical reaction. This is how you could have a de novo formation of membranes.”
They created the synthetic membranes from a watery emulsion of an oil and a detergent. Alone it’s stable. Add copper ions and sturdy vesicles and tubules begin to bud off the oil droplets. After 24 hours, the oil droplets are gone, “consumed” by the self-assembling membranes.
Although other scientists recently announced the creation of a “synthetic cell,” only its genome was artificial. The rest was a hijacked bacterial cell. Fully artificial life will require the union of both an information-carrying genome and a three-dimensional structure to house it.
The real value of this discovery might reside in its simplicity. From commercially available precursors, the scientists needed just one preparatory step to create each starting lipid chain.
“It’s trivial and can be done in a day,” Devaraj said. “New people who join the lab can make membranes from day one.”
Provided by University of California - San Diego (news : web)
Monday, February 13. 2012
Computer Algorithm Used To Make Movie For Sundance Film Festival
Wednesday, February 01. 2012
Killed by Code: Software Transparency in Implantable Medical Devices
Via Software Freedom Law Center
-----
Copyright © 2010, Software Freedom Law Center. Verbatim copying of this document is permitted in any medium; this notice must be preserved on all copies. †
I Abstract
Software is an integral component of a range of devices that perform critical, lifesaving functions and basic daily tasks. As patients grow more reliant on computerized devices, the dependability of software is a life-or-death issue. The need to address software vulnerability is especially pressing for Implantable Medical Devices (IMDs), which are commonly used by millions of patients to treat chronic heart conditions, epilepsy, diabetes, obesity, and even depression.
The software on these devices performs life-sustaining functions such as cardiac pacing and defibrillation, drug delivery, and insulin administration. It is also responsible for monitoring, recording and storing private patient information, communicating wirelessly with other computers, and responding to changes in doctors’ orders.
The Food and Drug Administration (FDA) is responsible for evaluating the risks of new devices and monitoring the safety and efficacy of those currently on market. However, the agency is unlikely to scrutinize the software operating on devices during any phase of the regulatory process unless a model that has already been surgically implanted repeatedly malfunctions or is recalled.
The FDA has issued 23 recalls of defective devices during the first half of 2010, all of which are categorized as “Class I,” meaning there is “reasonable probability that use of these products will cause serious adverse health consequences or death.” At least six of the recalls were likely caused by software defects.1 Physio-Control, Inc., a wholly owned subsidiary of Medtronic and the manufacturer of one defibrillator that was probably recalled due to software-related failures, admitted in a press release that it had received reports of similar failures from patients “over the eight year life of the product,” including one “unconfirmed adverse patient event.”2
Despite the crucial importance of these devices and the absence of comprehensive federal oversight, medical device software is considered the exclusive property of its manufacturers, meaning neither patients nor their doctors are permitted to access their IMD’s source code or test its security.3
In 2008, the Supreme Court of the United States’ ruling in Riegel v. Medtronic, Inc. made people with IMDs even more vulnerable to negligence on the part of device manufacturers.4 Following a wave of high-profile recalls of defective IMDs in 2005, the Court’s decision prohibited patients harmed by defects in FDA-approved devices from seeking damages against manufacturers in state court and eliminated the only consumer safeguard protecting patients from potentially fatal IMD malfunctions: product liability lawsuits. Prevented from recovering compensation from IMD-manufacturers for injuries, lost wages, or health expenses in the wake of device failures, people with chronic medical conditions are now faced with a stark choice: trust manufacturers entirely or risk their lives by opting against life-saving treatment.
We at the Software Freedom Law Center (SFLC) propose an unexplored solution to the software liability issues that are increasingly pressing as the population of IMD-users grows--requiring medical device manufacturers to make IMD source-code publicly auditable. As a non-profit legal services organization for Free and Open Source (FOSS) software developers, part of the SFLC’s mission is to promote the use of open, auditable source code5 in all computerized technology. This paper demonstrates why increased transparency in the field of medical device software is in the public’s interest. It unifies various research into the privacy and security risks of medical device software and the benefits of published systems over closed, proprietary alternatives. Our intention is to demonstrate that auditable medical device software would mitigate the privacy and security risks in IMDs by reducing the occurrence of source code bugs and the potential for malicious device hacking in the long-term. Although there is no way to eliminate software vulnerabilities entirely, this paper demonstrates that free and open source medical device software would improve the safety of patients with IMDs, increase the accountability of device manufacturers, and address some of the legal and regulatory constraints of the current regime.
We focus specifically on the security and privacy risks of implantable medical devices, specifically pacemakers and implantable cardioverter defibrillators, but they are a microcosm of the wider software liability issues which must be addressed as we become more dependent on embedded systems and devices. The broader objective of our research is to debunk the “security through obscurity” misconception by showing that vulnerabilities are spotted and fixed faster in FOSS programs compared to proprietary alternatives. The argument for public access to source code of IMDs can, and should be, extended to all the software people interact with everyday. The well-documented recent incidents of software malfunctions in voting booths, cars, commercial airlines, and financial markets are just the beginning of a problem that can only be addressed by requiring the use of open, auditable source code in safety-critical computerized devices.6
In section one, we give an overview of research related to potentially fatal software vulnerabilities in IMDs and cases of confirmed device failures linked to source code vulnerabilities. In section two, we summarize research on the security benefits of FOSS compared to closed-source, proprietary programs. In section three, we assess the technical and legal limitations of the existing medical device review process and evaluate the FDA’s capacity to assess software security. We conclude with our recommendations on how to promote the use of FOSS in IMDs. The research suggests that the occurrence of privacy and security breaches linked to software vulnerabilities is likely to increase in the future as embedded devices become more common.
II Software Vulnerabilities in IMDs
A variety of wirelessly reprogrammable IMDs are surgically implanted directly into the body to detect and treat chronic health conditions. For example, an implantable cardioverter defibrillator, roughly the size of a small mobile phone, connects to a patient’s heart, monitors rhythm, and delivers an electric shock when it detects abnormal patterns. Once an IMD has been implanted, health care practitioners extract data, such as electrocardiogram readings, and modify device settings remotely, without invasive surgery. New generation ICDs can be contacted and reprogrammed via wireless radio signals using an external device called a “programmer.”
In 2008, approximately 350,000 pacemakers and 140,000 ICDs were implanted in the United States, according to a forecast on the implantable medical device market published earlier this year.7 Nation-wide demand for all IMDs is projected to increase 8.3 percent annually to $48 billion by 2014, the report says, as “improvements in safety and performance properties …enable ICDs to recapture growth opportunities lost over the past year to product recall.”8 Cardiac implants in the U.S. will increase 7.3 percent annually, the article predicts, to $16.7 billion in 2014, and pacing devices will remain the top-selling group in this category.9
Though the surge in IMD treatment over the past decade has had undeniable health benefits, device failures have also had fatal consequences. From 1997 to 2003, an estimated 400,000 to 450,000 ICDs were implanted world-wide and the majority of the procedures took place in the United States. At least 212 deaths from device failures in five different brands of ICD occurred during this period, according to a study of the adverse events reported to the FDA conducted by cardiologists from the Minneapolis Heart Institute Foundation.10
Research indicates that as IMD usage grows, the frequency of potentially fatal software glitches, accidental device malfunctions, and the possibility of malicious attacks will grow. While there has yet to be a documented incident in which the source code of a medical device was breached for malicious purposes, a 2008-study led by software engineer and security expert Kevin Fu proved that it is possible to interfere with an ICD that had passed the FDA’s premarket approval process and been implanted in hundreds of thousands of patients. A team of researchers from three universities partially reverse-engineered the communications protocol of a 2003-model ICD and launched several radio-based software attacks from a short distance. Using low-cost, commercially available equipment to bypass the device programmer, the researchers were able to extract private data stored inside the ICD such as patients’ vital signs and medical history; “eavesdrop” on wireless communication with the device programmer; reprogram the therapy settings that detect and treat abnormal heart rhythms; and keep the device in “awake” mode in order to deplete its battery, which can only be replaced with invasive surgery.
In one experimental attack conducted in the study, researchers were able to first disable the ICD to prevent it from delivering a life-saving shock and then direct the same device to deliver multiple shocks averaging 137.7 volts that would induce ventricular fibrillation in a patient. The study concluded that there were no “technological mechanisms in place to ensure that programmers can only be operated by authorized personnel.” Fu’s findings show that almost anyone could use store-bought tools to build a device that could “be easily miniaturized to the size of an iPhone and carried through a crowded mall or subway, sending its heart-attack command to random victims.”11
The vulnerabilities Fu’s lab exploited are the result of the very same features that enable the ICD to save lives. The model breached in the experiment was designed to immediately respond to reprogramming instructions from health-care practitioners, but is not equipped to distinguish whether treatment requests originate from a doctor or an adversary. An earlier paper co-authored by Fu proposed a solution to the communication-security paradox. The paper recommends the development of a wearable “cloaker” for IMD-patients that would prevent anyone but pre-specified, authorized commercial programmers to interact with the device. In an emergency situation, a doctor with a previously unauthorized commercial programmer would be able to enable emergency access to the IMD by physically removing the cloaker from the patient.12
Though the adversarial conditions demonstrated in Fu’s studies were hypothetical, two early incidents of malicious hacking underscore the need to address the threat software liabilities pose to the security of IMDs. In November 2007, a group of attackers infiltrated the Coping with Epilepsy website and planted flashing computer animations that triggered migraine headaches and seizures in photosensitive site visitors.13 A year later, malicious hackers mounted a similar attack on the Epilepsy Foundation website.14
Ample evidence of software vulnerabilities in other common IMD treatments also indicates that the worst-case scenario envisioned by Fu’s research is not unfounded. From 1983 to 1997 there were 2,792 quality problems that resulted in recalls of medical devices, 383 of which were related to computer software, according to a 2001 study analyzing FDA reports of the medical devices that were voluntarily recalled by manufacturers over a 15-year period.15 Cardiology devices accounted for 21 percent of the IMDs that were recalled. Authors Dolores R. Wallace and D. Richard Kuhn discovered that 98 percent of the software failures they analyzed would have been detected through basic scientific testing methods. While none of the failures they researched caused serious personal injury or death, the paper notes that there was not enough information to determine the potential health consequences had the IMDs remained in service.
Nearly 30 percent of the software-related recalls investigated in the report occurred between 1994 and 1996. “One possibility for this higher percentage in later years may be the rapid increase of software in medical devices. The amount of software in general consumer products is doubling every two to three years,” the report said.
As more individual IMDs are designed to automatically communicate wirelessly with physician’s offices, hospitals, and manufacturers, routine tasks like reprogramming, data extraction, and software updates may spur even more accidental software glitches that could compromise the security of IMDs.
The FDA launched an “Infusion Pump Improvement Initiative” in April 2010, after receiving thousands of reports of problems associated with the use of infusion pumps that deliver fluids such as insulin and chemotherapy medication to patients electronically and mechanically.16 Between 2005 and 2009, the FDA received approximately 56,000 reports of adverse events related to infusion pumps, including numerous cases of injury and death. The agency analyzed the reports it received during the period in a white paper published in the spring of 2010 and found that the most common types of problems reported were associated with software defects or malfunctions, user interface issues, and mechanical or electrical failures. (The FDA said most of the pumps covered in the report are operated by a trained user, who programs the rate and duration of fluid delivery through a built-in software interface). During the period, 87 infusion pumps were recalled due to safety concerns, 14 of which were characterized as “Class I” – situations in which there is a reasonable probability that use of the recalled device will cause serious adverse health consequences or death. Software defects lead to over-and-under infusion and caused pre-programmed alarms on pumps to fail in emergencies or activate in absence of a problem. In one instance a “key bounce” caused an infusion pump to occasionally register one keystroke (e.g., a single zero, “0”) as multiple keystrokes (e.g., a double zero, “00”), causing an inappropriate dosage to be delivered to a patient. Though the report does not apply to implantable infusion pumps, it demonstrates the prevalence of software-related malfunctions in medical device software and the flexibility of the FDA’s pre-market approval process.
In order to facilitate the early detection and correction of any design defects, the FDA has begun offering manufacturers “the option of submitting the software code used in their infusion pumps for analysis by agency experts prior to premarket review of new or modified devices.” It is also working with third-party researchers to develop “an open-source software safety model and reference specifications that infusion pump manufacturers can use or adapt to verify the software in their devices.”
Though the voluntary initiative appears to be an endorsement of the safety benefits of FOSS and a step in the right direction, it does not address the overall problem of software insecurity since it is not mandatory.
III Why Free Software is More Secure
“Continuous and broad peer-review, enabled by publicly available source code, improves software reliability and security through the identification and elimination of defects that might otherwise go unrecognized by the core development team. Conversely, where source code is hidden from the public, attackers can attack the software anyway …. Hiding source code does inhibit the ability of third parties to respond to vulnerabilities (because changing software is more difficult without the source code), but this is obviously not a security advantage. In general, ‘Security by Obscurity’ is widely denigrated.” — Department of Defense (DoD) FAQ’s response to question: “Doesn’t Hiding Source Code Automatically Make Software More Secure?”17
The DoD indicates that FOSS has been central to its Information Technology (IT) operations since the mid-1990’s, and, according to some estimates, one-third to one-half of the software currently used by the agency is open source.18 The U.S. Office of Management and Budget issued a memorandum in 2004, which recommends that all federal agencies use the same procurement procedures for FOSS as they would for proprietary software.19 Other public sector agencies, such as the U.S. Navy, the Federal Aviation Administration, the U.S. Census Bureau and the U.S. Patent and Trademark Office have been identified as recognizing the security benefits of publicly auditable source code.20
To understand why free and open source software has become a common component in the IT systems of so many businesses and organizations that perform life-critical or mission-critical functions, one must first accept that software bugs are a fact of life. The Software Engineering Institute estimates that an experienced software engineer produces approximately one defect for every 100 lines of code.21 Based on this estimate, even if most of the bugs in a modest, one million-line code base are fixed over the course of a typical program life cycle, approximately 1,000 bugs would remain.
In its first “State of Software Security” report released in March 2010, the private software security analysis firm Veracode reviewed the source code of 1,591 software applications voluntarily submitted by commercial vendors, businesses, and government agencies.22 Regardless of program origins, Veracode found that 58 percent of all software submitted for review did not meet the security assessment criteria the report established.23 Based on its findings, Veracode concluded that “most software is indeed very insecure …[and] more than half of the software deployed in enterprises today is potentially susceptible to an application layer attack similar to that used in the recent …Google security breaches.”24
Though open source applications had almost as many source code vulnerabilities upon first submission as proprietary programs, researchers found that they contained fewer potential backdoors than commercial or outsourced software and that open source project teams remediated security vulnerabilities within an average of 36 days of the first submission, compared to 48 days for internally developed applications and 82 days for commercial applications.25 Not only were bugs patched the fastest in open source programs, but the quality of remediation was also higher than commercial programs.26
Veracode’s study confirms the research and anecdotal evidence into the security benefits of open source software published over the past decade. According to the web-security analysis site SecurityPortal, vulnerabilities took an average of 11.2 days to be spotted in Red Hat/Linux systems with a standard deviation of 17.5 compared to an average of 16.1 days with a standard deviation of 27.7 in Microsoft programs.27
Sun Microsystems’ COO Bill Vass summed up the most common case for FOSS in a blog post published in April 2009: “By making the code open source, nothing can be hidden in the code,” Vass wrote. “If the Trojan Horse was made of glass, would the Trojans have rolled it into their city? NO.”28
Vass’ logic is backed up by numerous research papers and academic studies that have debunked the myth of security through obscurity and advanced the “more eyes, fewer bugs” thesis. Though it might seem counterintuitive, making source code publicly available for users, security analysts, and even potential adversaries does not make systems more vulnerable to attack in the long-run. To the contrary, keeping source code under lock-and-key is more likely to hamstring “defenders” by preventing them from finding and patching bugs that could be exploited by potential attackers to gain entry into a given code base, whether or not access is restricted by the supplier.29 “In a world of rapid communications among attackers where exploits are spread on the Internet, a vulnerability known to one attacker is rapidly learned by others,” reads a 2006 article comparing open source and proprietary software use in government systems.30 “For Open Source, the next assumption is that disclosure of a flaw will prompt other programmers to improve the design of defenses. In addition, disclosure will prompt many third parties — all of those using the software or the system — to install patches or otherwise protect themselves against the newly announced vulnerability. In sum, disclosure does not help attackers much but is highly valuable to the defenders who create new code and install it.”
Academia and internet security professionals appear to have reached a consensus that open, auditable source code gives users the ability to independently assess the exposure of a system and the risks associated with using it; enables bugs to be patched more easily and quickly; and removes dependence on a single party, forcing software suppliers and developers to spend more effort on the quality of their code, as authors Jaap-Henk Hoepman and Bart Jacobs also conclude in their 2007 article, Increased Security Through Open Source.31
By contrast, vulnerabilities often go unnoticed, unannounced, and unfixed in closed source programs because the vendor, rather than users who have a higher stake in maintaining the quality of software, is the only party allowed to evaluate the security of the code base.32 Some studies have argued that commercial software suppliers have less of an incentive to fix defects after a program is initially released so users do not become aware of vulnerabilities until after they have caused a problem. “Once the initial version of [a proprietary software product] has saturated its market, the producer’s interest tends to shift to generating upgrades …Security is difficult to market in this process because, although features are visible, security functions tend to be invisible during normal operations and only visible when security trouble occurs.”33
The consequences of manufacturers’ failure to disclose malfunctions to patients and physicians have proven fatal in the past. In 2005, a 21-year-old man died from cardiac arrest after the ICD he wore short-circuited and failed to deliver a life-saving shock. The fatal incident prompted Guidant, the manufacturer of the flawed ICD, to recall four different device models they sold. In total 70,000 Guidant ICDs were recalled in one of the biggest regulatory actions of the past 25 years.34
Guidant came under intense public scrutiny when the patient’s physician Dr. Robert Hauser discovered that the company first observed the flaw that caused his patient’s device to malfunction in 2002, and even went so far as to implement manufacturing changes to correct it, but failed to disclose it to the public or health-care industry.
The body of research analyzed for this paper points to the same conclusion: security is not achieved through obscurity and closed source programs force users to forfeit their ability to evaluate and improve a system’s security. Though there is lingering debate over the degree to which end-users contribute to the maintenance of FOSS programs and how to ensure the quality of the patches submitted, most of the evidence supports our paper’s central assumption that auditable, peer-reviewed software is comparatively more secure than proprietary programs.
Programs have different standards to ensure the quality of the patches submitted to open source programs, but even the most open, transparent systems have established methods of quality control. Well-established open source software, such as the kind favored by the DoD and the other agencies mentioned above, cannot be infiltrated by “just anyone.” To protect the code base from potential adversaries and malicious patch submissions, large open source systems have a “trusted repository” that only certain, “trusted,” developers can directly modify. As an additional safeguard, the source code is publicly released, meaning not only are there more people policing it for defects, but more copies of each version of the software exist making it easier to compare new code.
IV The FDA Review Process and Legal Obstacles to Device Manufacturer Accountability
“Implanted medical devices have enriched and extended the lives of countless people, but device malfunctions and software glitches have become modern ‘diseases’ that will continue to occur. The failure of manufacturers and the FDA to provide the public with timely, critical information about device performance, malfunctions, and ’fixes’ enables potentially defective devices to reach unwary consumers.” — Capitol Hill Hearing Testimony of William H. Maisel, Director of Beth Israel Deaconess Medical Center, May 12, 2009.
The FDA’s Center for Devices and Radiological Health (CDRH) is responsible for regulating medical devices, but since the Medical Device Modernization Act (MDMA) was passed in 1997 the agency has increasingly ceded control over the pre-market evaluation and post-market surveillance of IMDs to their manufacturers.35 While the agency has been making strides towards the MDMA’s stated objective of streamlining the device approval process, the expedient regulatory process appears to have come at the expense of the CDRH’s broader mandate to “protect the public health in the fields of medical devices” by developing and implementing programs “intended to assure the safety, effectiveness, and proper labeling of medical devices.”36
Since the MDMA was passed, the FDA has largely deferred these responsibilities to the companies that sell these devices.37 The legislation effectively allowed the businesses selling critical devices to establish their own set of pre-market safety evaluation standards and determine the testing conducted during the review process. Manufacturers also maintain a large degree of discretion over post-market IMD surveillance. Though IMD-manufacturers are obligated to inform the FDA if they alert physicians to a product defect or if the device is recalled, they determine whether a particular defect constitutes a public safety risk.
“Manufacturers should use good judgment when developing their quality system and apply those sections of the QS regulation that are applicable to their specific products and operations,” reads section 21 of Quality Standards regulation outlined on the FDA’s website. “Operating within this flexibility, it is the responsibility of each manufacturer to establish requirements for each type or family of devices that will result in devices that are safe and effective, and to establish methods and procedures to design, produce, distribute, etc. devices that meet the quality system requirements. The responsibility for meeting these requirements and for having objective evidence of meeting these requirements may not be delegated even though the actual work may be delegated.”
By implementing guidelines such as these, the FDA has refocused regulation from developing and implementing programs in the field of medical devices that protect the public health to auditing manufacturers’ compliance with their own standards.
“To the FDA, you are the experts in your device and your quality programs,” Jeff Geisler wrote in a 2010 book chapter, Software for Medical Systems.38 “Writing down the procedures is necessary — it is assumed that you know best what the procedures should be — but it is essential that you comply with your written procedures.”
The elastic regulatory standards are a product of the 1976 amendment to the Food, Drug, and Cosmetics Act.39The Amendment established three different device classes and outlined broad pre-market requirements that each category of IMD must meet depending on the risk it poses to a patient. Class I devices, whose failure would have no adverse health consequences, are not subject to a pre-market approval process. Class III devices that “support or sustain human life, are of substantial importance in preventing impairment of human health, or which present a potential, unreasonable risk of illness or injury,” such as IMDs, are subject to the most stringent FDA review process, Premarket Approval (PMA).40
By the FDA’s own admission, the original legislation did not account for technological complexity of IMDs, but neither has subsequent regulation.41
In 2002, an amendment to the MDMA was passed that was intended to help the FDA “focus its limited inspection resources on higher-risk inspections and give medical device firms that operate in global markets an opportunity to more efficiently schedule multiple inspections,” the agency’s website reads.42
The legislation further eroded the CDRH’s control over pre-market approval data, along with the FDA’s capacity to respond to rapid changes in medical treatment and the introduction of increasingly complex devices for a broader range of diseases. The new provisions allowed manufacturers to pay certain FDA-accredited inspectors to conduct reviews during the PMA process in lieu of government regulators. It did not outline specific software review procedures for the agency to conduct or precise requirements that medical device manufacturers must meet before introducing a new product. “The regulation …provides some guidance [on how to ensure the reliability of medical device software],” Joe Bremner wrote of the FDA’s guidance on software validation.43 “Written in broad terms, it can apply to all medical device manufacturers. However, while it identifies problems to be solved or an end point to be achieved, it does not specify how to do so to meet the intent of the regulatory requirement.”
The death of 21-year-old Joshua Oukrop in 2005 due to the failure of a Guidant device has increased calls for regulatory reform at the FDA.44 In a paper published shortly after Oukrop’s death, his physician, Dr. Hauser concluded that the FDA’s post-market ICD device surveillance system is broken.45
After returning the failed Prizm 2 DR pacemaker to Guidant, Dr. Hauser learned that the company had known the model was prone to the same defect that killed Oukrop for at least three years. Since 2002, Guidant received 25 different reports of failures in the Prizm model, three of which required rescue defibrillation. Though the company was sufficiently concerned about the problem to make manufacturing changes, Guidant continued to sell earlier models and failed to make patients and physicians aware that the Prizm was prone to electronic defects. They claimed that disclosing the defect was inadvisable because the risk of infection during de-implantation surgery posed a greater threat to public safety than device failure. “Guidant’s statistical argument ignored the basic tenet that patients have a fundamental right to be fully informed when they are exposed to the risk of death no matter how low that risk may be perceived,” Dr. Hauser argued. “Furthermore, by withholding vital information, Guidant had in effect assumed the primary role of managing high-risk patients, a responsibility that belongs to physicians. The prognosis of our young, otherwise healthy patient for a long, productive life was favorable if sudden death could have been prevented.”
The FDA was also guilty of violating the principle of informed consent. In 2004, Guidant had reported two different adverse events to the FDA that described the same defect in the Prizm 2 DR models, but the agency also withheld the information from the public. “The present experience suggests that the FDA is currently unable to satisfy its legal responsibility to monitor the safety of market released medical devices like the Prizm 2 DR,” he wrote, referring to the device whose failure resulted in his patient’s death. “The explanation for the FDA’s inaction is unknown, but it may be that the agency was not prepared for the extraordinary upsurge in ICD technology and the extraordinary growth in the number of implantations that has occurred in the past five years.”
While the Guidant recalls prompted increased scrutiny on the FDA’s medical device review process, it remains difficult to gauge the precise process of regulating IMD software or the public health risk posed by source code bugs since neither doctors, nor IMD users, are permitted to access it. Nonetheless, the information that does exist suggests that the pre-market approval process alone is not a sufficient consumer safeguard since medical devices are less likely than drugs to have demonstrated clinical safety before they are marketed.46
An article published in the Journal of the American Medical Association (JAMA) studied the safety and effectiveness data in every PMA application the FDA reviewed from January 2000 to December 2007 and concluded that “premarket approval of cardiovascular devices by the FDA is often based on studies that lack strength and may be prone to bias.”47 Of the 78 high-risk device approvals analyzed in the paper, 51 were based on a single study.48
The JAMA study noted that the need to address the inadequacies of the FDA’s device regulation process has become particularly urgent since the Supreme Court changed the landscape of medical liability law with its ruling in Riegel v. Medtronic in February 2008. The Court held that the plaintiff Charles Riegel could not seek damages in state court from the manufacturer of a catheter that exploded in his leg during an angioplasty. “Riegel v. Medtronic means that FDA approval of a device preempts consumers from suing because of problems with the safety or effectiveness of the device, making this approval a vital consumer protection safeguard.”49
Since the FDA is a federal agency, its authority supersedes state law. Based on the concept of preemption, the Supreme Court held that damages actions permitted under state tort law could not be filed against device manufacturers deemed to be in compliance with the FDA, even in the event of gross negligence. The decision eroded one of the last legal recourses to protect consumers and hold IMD manufacturers accountable for catastrophic, failure of an IMD. Not only are the millions of people who rely on IMD’s for their most life-sustaining bodily functions more vulnerable to software malfunctions than ever before, but they have little choice but to trust its manufacturers.
“It is clear that medical device manufacturers have responsibilities that extend far beyond FDA approval and that many companies have failed to meet their obligations,” William H. Maisel said in recent congressional testimony on the Medical Device Reform bill.50 “Yet, the U.S. Supreme Court ruled in their February 2008 decision, Riegel v. Medtronic, that manufacturers could not be sued under state law by patients harmed by product defects from FDA-approved medical devices …. [C]onsumers are unable to seek compensation from manufacturers for their injuries, lost wages, or health expenses. Most importantly, the Riegel decision eliminates an important consumer safeguard — the threat of manufacturer liability — and will lead to less safe medical devices and an increased number of patient injuries.”
In light of our research and the existing legal and regulatory limitations that prevent IMD users from holding medical device manufacturers accountable for critical software vulnerabilities, auditable source code is critical to minimize the harm caused by inevitable medical device software bugs.
V Conclusion
The Supreme Court’s decision in favor of Medtronic in 2008, increasingly flexible regulation of medical device software on the part of the FDA, and a spike in the level and scope of IMD usage over the past decade suggest a software liability nightmare on the horizon. We urge the FDA to introduce more stringent, mandatory standards to protect IMD-wearers from the potential adverse consequences of software malfunctions discussed in this paper. Specifically, we call on the FDA to require manufacturers of life-critical IMDs to publish the source code of medical device software so the public and regulators can examine and evaluate it. At the very least, we urge the FDA to establish a repository of medical device software running on implanted IMDs in order to ensure continued access to source code in the event of a catastrophic failure, such as the bankruptcy of a device manufacturer. While we hope regulators will require that the software of all medical devices, regardless of risk, be auditable, it is particularly urgent that these standards be included in the pre-market approval process of Class III IMDs. We hope this paper will also prompt IMD users, their physicians, and the public to demand greater transparency in the field of medical device software.
Notes
1Though software defects are never explicitly mentioned as the “Reason for Recall” in the alerts posted on the FDA’s website, the descriptions of device failures match those associated with souce-code errors. List of Device Recalls, U.S. Food & Drug Admin., http://www.fda.gov/MedicalDevices/Safety/RecallsCorrectionsRemovals/ListofRecalls/default.htm (last visited Jul. 19, 2010).
2Medtronic recalled its Lifepak 15 Monitor/Defibrillator in March 4, 2010 due to failures that were almost certainly caused by software defects that caused the device to unexpectedly shut-down and regain power on its own. The company admitted in a press release that it first learned that the recalled model was prone to defects eight years earlier and had submitted one “adverse event” report to the FDA. Medtronic, Physio-Control Field Correction to LIFEPAK 20/20e Defibrillator/ Monitors, BusinessWire (Jul. 02, 2010, 9:00 AM), http://www.businesswire.com/news/home/20100702005034/en/Physio-Control-Field-Correction-LIFEPAK%C2%AE-2020e-Defibrillator-Monitors.html
20/20e Defibrillator/ Monitors, BusinessWire (Jul. 02, 2010, 9:00 AM), http://www.businesswire.com/news/home/20100702005034/en/Physio-Control-Field-Correction-LIFEPAK%C2%AE-2020e-Defibrillator-Monitors.html
3Quality Systems Regulation, U.S. Food & Drug Admin., http://www.fda.gov/MedicalDevices/default.htm (follow “Device Advice: Device Regulation and Guidance” hyperlink; then follow “Postmarket Requirements (Devices)” hyperlink; then follow “Quality Systems Regulation” hyperlink) (last visited Jul. 2010)
4Riegel v. Medtronic, Inc., 552 U.S. 312 (2008).
5The Software Freedom Law Center (SFLC) prefers the term Free and Open Source Software (FOSS) to describe software that can be freely viewed, used, and modified by anyone. In this paper, we sometimes use mixed terminology, including the term “open source” to maintain consistency with the articles cited.
6See Josie Garthwaite, Hacking the Car: Cyber Security Risks Hit the Road, Earth2Tech (Mar. 19, 2010, 12:00 AM), http://earth2tech.com.
7Sanket S. Dhruva et al., Strength of Study Evidence Examined by the FDA in Premarket Approval of Cardiovascular Devices, 302 J. Am. Med. Ass’n 2679 (2009).
8Freedonia Group, Cardiac Implants, Rep. Buyer, Sept. 2008, available at
http://www.reportbuyer.com/pharma_healthcare/medical_devices/cardiac_implants.html.
9Id.
10Robert G. Hauser & Linda Kallinen, Deaths Associated With Implantable Cardioverter Defibrillator Failure and Deactivation Reported in the United States Food and Drug Administration Manufacturer and User Facility Device Experience Database, 1 HeartRhythm 399, available at http://www.heartrhythmjournal.com/article/S1547-5271%2804%2900286-3/.
11Charles Graeber, Profile of Kevin Fu, 33, TR35 2009 Innovator, Tech. Rev.,
http://www.technologyreview.com/TR35/Profile.aspx?trid=760 (last visited Jul. 9, 2010).
12See Tamara Denning, et al., Absence Makes the Heart Grow Fonder: New Directions for Implantable Medical Device Security, Proceedings of the 3rd Conference on Hot Topics in Security(2008), available at
http://www.cs.washington.edu/homes/yoshi/papers/HotSec2008/cloaker-hotsec08.pdf.
13Kevin Poulsen, Hackers Assault Epilepsy Patients via Computer, Wired News (Mar. 28, 2008),
http://www.wired.com/politics/security/news/2008/03/epilepsy.
14Id.
15Dolores R. Wallace & D. Richard Kuhn, Failure Modes in Medical Device Software: An Analysis of 15 Years of Recall Data, 8 Int’l J. Reliability Quality Safety Eng’g 351 (2001), available at http://csrc.nist.gov/groups/SNS/acts/documents/final-rqse.pdf.
Wednesday, January 25. 2012
Google’s Broken Promise: The End of "Don’t Be Evil"
Tuesday, January 24. 2012
A tale of Apple, the iPhone, and overseas manufacturing
Via CNET
-----

Workers assemble and perform quality control checks on MacBook Pro display enclosures at an Apple supplier facility in Shanghai.
(Credit: Apple)
A new report on Apple offers up an interesting detail about the evolution of the iPhone and gives a fascinating--and unsettling--look at the practice of overseas manufacturing.
The article, an in-depth report by Charles Duhigg and Keith Bradsher of The New York Times, is based on interviews with, among others, "more than three dozen current and former Apple employees and contractors--many of whom requested anonymity to protect their jobs."
The piece uses Apple and its recent history to look at why the success of some U.S. firms hasn't led to more U.S. jobs--and to examine issues regarding the relationship between corporate America and Americans (as well as people overseas). One of the questions it asks is: Why isn't more manufacturing taking place in the U.S.? And Apple's answer--and the answer one might get from many U.S. companies--appears to be that it's simply no longer possible to compete by relying on domestic factories and the ecosystem that surrounds them.
The iPhone detail crops up relatively early in the story, in an anecdote about then-Apple CEO Steve Jobs. And it leads directly into questions about offshore labor practices:
In 2007, a little over a month before the iPhone was scheduled to appear in stores, Mr. Jobs beckoned a handful of lieutenants into an office. For weeks, he had been carrying a prototype of the device in his pocket.
Mr. Jobs angrily held up his iPhone, angling it so everyone could see the dozens of tiny scratches marring its plastic screen, according to someone who attended the meeting. He then pulled his keys from his jeans.
People will carry this phone in their pocket, he said. People also carry their keys in their pocket. "I won't sell a product that gets scratched," he said tensely. The only solution was using unscratchable glass instead. "I want a glass screen, and I want it perfect in six weeks."
A tall order. And another anecdote suggests that Jobs' staff went overseas to fill it--along with other requirements for the top-secret phone project (code-named, the Times says, "Purple 2"):
One former executive described how the company relied upon a Chinese factory to revamp iPhone manufacturing just weeks before the device was due on shelves. Apple had redesigned the iPhone's screen at the last minute, forcing an assembly line overhaul. New screens began arriving at the plant near midnight.
A foreman immediately roused 8,000 workers inside the company's dormitories, according to the executive. Each employee was given a biscuit and a cup of tea, guided to a workstation and within half an hour started a 12-hour shift fitting glass screens into beveled frames. Within 96 hours, the plant was producing more than 10,000 iPhones a day.
"The speed and flexibility is breathtaking," the executive said. "There's no American plant that can match that."
That last quote there, like several others in the story, leaves one feeling almost impressed by the no-holds-barred capabilities of these manufacturing plants--impressed and queasy at the same time. Here's another quote, from Jennifer Rigoni, Apple's worldwide supply demand manager until 2010: "They could hire 3,000 people overnight," she says, speaking of Foxconn City, Foxconn Technology's complex of factories in China. "What U.S. plant can find 3,000 people overnight and convince them to live in dorms?"
The article says that cheap and willing labor was indeed a factor in Apple's decision, in the early 2000s, to follow most other electronics companies in moving manufacturing overseas. But, it says, supply chain management, production speed, and flexibility were bigger incentives.
"The entire supply chain is in China now," the article quotes a former high-ranking Apple executive as saying. "You need a thousand rubber gaskets? That's the factory next door. You need a million screws? That factory is a block away. You need that screw made a little bit different? It will take three hours."
It also makes the point that other factors come into play. Apple analysts, the Times piece reports, had estimated that in the U.S., it would take the company as long as nine months to find the 8,700 industrial engineers it would need to oversee workers assembling the iPhone. In China it wound up taking 15 days.
The article and its sources paint a vivid picture of how much easier it is for companies to get things made overseas (which is why so many U.S. firms go that route--Apple is by no means alone in this). But the underlying humanitarian issues nag at the reader.
Perhaps there's hope--at least for overseas workers--in last week's news that Apple has joined the Fair Labor Association, and that it will be providing more transparency when it comes to the making of its products.
As for manufacturing returning to the U.S.? The Times piece cites an unnamed guest at President Obama's 2011 dinner with Silicon Valley bigwigs. Obama had asked Steve Jobs what it would take to produce the iPhone in the states, why that work couldn't return. The Times' source quotes Jobs as having said, in no uncertain terms, "Those jobs aren't coming back."
Apple, by the way, would not provide a comment to the Times about the article. And Foxconn disputed the story about employees being awakened at midnight to work on the iPhone, saying strict regulations about working hours would have made such a thing impossible.
Quicksearch
Popular Entries
- The great Ars Android interface shootout (131506)
- Norton cyber crime study offers striking revenue loss statistics (102390)
- MeCam $49 flying camera concept follows you around, streams video to your phone (100517)
- The PC inside your phone: A guide to the system-on-a-chip (58691)
- Norton cyber crime study offers striking revenue loss statistics (58619)
Categories

Show tagged entries
Syndicate This Blog
Calendar
|
|
July '26 | |||||
| Mon | Tue | Wed | Thu | Fri | Sat | Sun |
| 1 | 2 | 3 | 4 | 5 | ||
| 6 | 7 | 8 | 9 | 10 | 11 | 12 |
| 13 | 14 | 15 | 16 | 17 | 18 | 19 |
| 20 | 21 | 22 | 23 | 24 | 25 | 26 |
| 27 | 28 | 29 | 30 | 31 | ||